DANVERS, Massachusetts–(BUSINESS WIRE)–Les données présentées lors du TCT Connect, 32e symposium scientifique annuel de la Cardiovascular Research Foundation, démontrent que l’identification précoce d’une insuffisance cardiaque droite et l’utilisation précoce de l’Impella RP favorisent un meilleur taux de survie. L’identification précoce des patients nécessitant une assistance cardiaque droite est essentielle car des études antérieures ont démontré que 37 % des patients ayant subi un choc cardiogénique suite à un IAM (AMICS) présentent un dysfonctionnement cardiaque droit 1, entraînant un risque de mortalité 2 huit fois plus élevé.
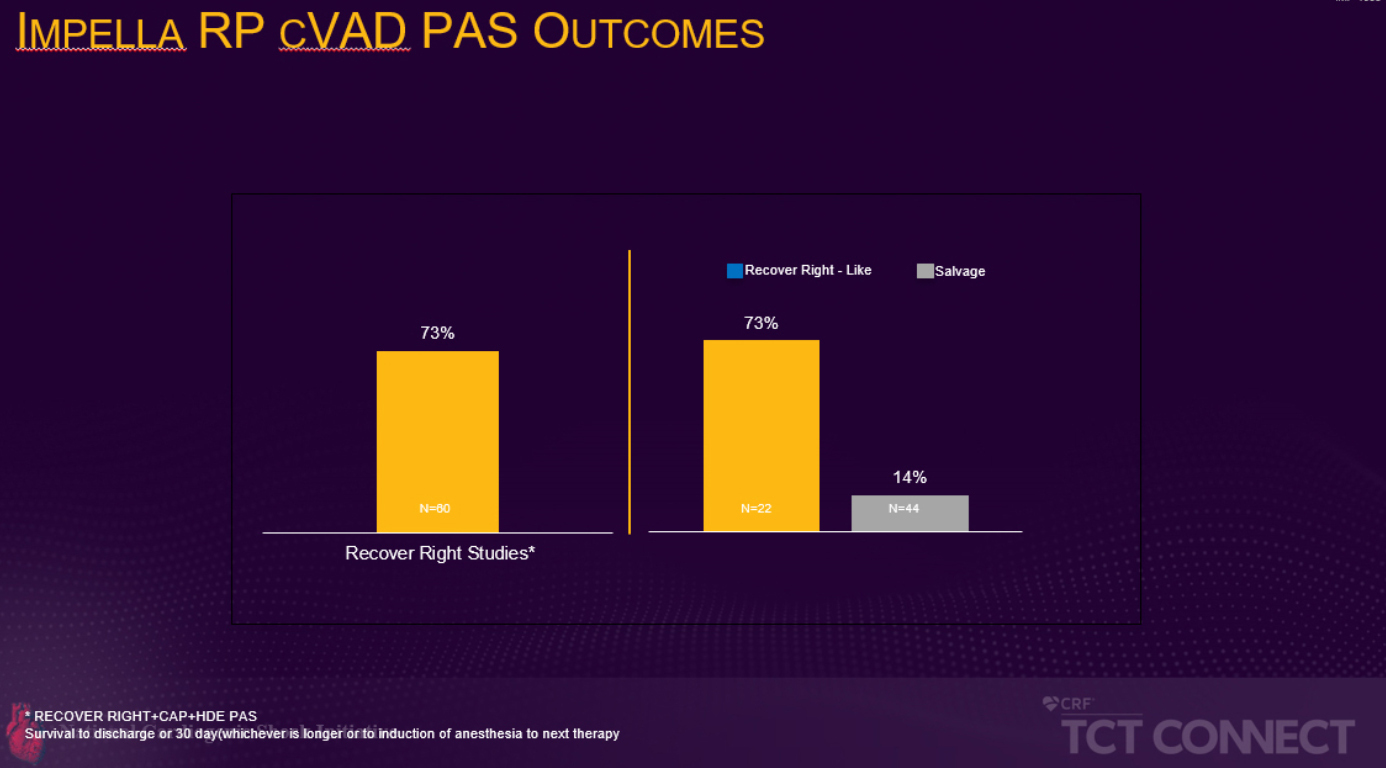

L’étude prospective, multicentrique, post approbation FDA en vue d’une autorisation PMA (autorisation préalable à la mise sur le marché) présentée lors du TCT a comparé la survie des patients qui auraient satisfait aux critères de recrutement pour l’essai RECOVER RIGHT à ceux qui n’auraient pas satisfait ces critères suite à un état de choc cardiogénique depuis plus de 48 heures. L’essai RECOVER RIGHT et les données subséquentes à l’étude post-approbation HDE (exemption pour motif humanitaire) ont été rassemblés entre 2012 et 2017 et ont conduit à une autorisation PMA pour l’Impella RP en 2017. L’étude post-approbation PMA en cours présentée lors du TCT Connect démontre que les patients enrôlés entre septembre 2017 et juin 2019 ayant reçu l’assistance de l’Impella RP dans les 48 heures suivant le début du choc cardiogénique connaissent un taux de survie significativement plus élevé que ceux recevant une assistance cardiaque droite tardive (73 % contre 14 %, p<0,001). Le taux de survie de 73 % est comparable au taux de survie des études pré-PMA RECOVER RIGHT et HDE. (voir figure 1)
« La détection précoce d’une insuffisance cardiaque droite et une intervention rapide sont essentielles à l’amélioration des taux de survie des patients », a déclaré le docteur Mark Anderson, président du département de chirurgie cardiaque du centre médical HUMC/Hackensack Meridian Health. « Cette étude suggère qu’en cas d’insuffisance cardiaque droite, le fait de minimiser le temps entre le début du choc et l’utilisation de l’Impella RP est essentiel et permet d’accompagner le patient de façon appropriée. »
Une deuxième étude présentée lors du TCT vise à accompagner les cliniciens dans l’identification précoce des facteurs déclenchants de l’insuffisance cardiaque droite. L’examen de 100 patients réalisé par des chercheurs chevronnés de l’étude National Cardiogenic Shock Initiative (NCSI), a comparé des patients atteints d’une insuffisance ventriculaire droite compliquée par un choc cardiogénique (AMICS) à d’autres patients qui n’en souffrent pas et a démontré qu’une dysfonction diastolique persistante sur la console automatisée Impella ainsi qu’une pression veineuse centrale élevée supérieure à 12 mmHg pouvait correspondre à une indication précoce d’insuffisance ventriculaire droite. (voir figure 2)
« Les informations en temps réel fournit par la console automatisée Impella sont un outil important pour les médecins afin qu’ils obtiennent de meilleurs résultats pour les patients », a déclaré Babar Basir, D.O., cardiologue interventionnel à l’hôpital Henry Ford. « Une dysfonction diastolique prolongée peut être un marqueur précoce d’insuffisance ventriculaire droite chez les patients présentant des pressions de remplissage élevées, et une dysfonction diastolique de plus longue durée entraîne une dégradation des résultats. »
L’Impella RP est le dispositif d’assistance du ventricule droit le plus étudié ainsi que la seule technologie percutanée ayant reçu l’autorisation de la FDA qui le désigne comme sûr et efficace en matière d’assistance ventriculaire droite. L’autorisation exceptionnelle émise par la FDA est le résultat de cinq années de recherche qui englobent :
- RECOVER RIGHT, une étude prospective, multicentrique et à groupe unique approuvée par la FDA, qui a débuté après que la société ait reçu l’autorisation IDE (exemption relative aux dispositifs expérimentaux) de la FDA en novembre 2012 et qui s’est achevée en 2014
- Un protocole d’accès continu
- Une étude post-approbation HDE, achevée en 2017
- Une étude post-approbation PMA, initiée en septembre 2017
De plus, le 29 mai 2020, la FDA a délivré une autorisation d’utilisation d’urgence (EUA) afin d’étendre l’utilisation de l’Impella RP aux patients souffrant de complications ventriculaire droite liées à la COVID-19, y compris un dysfonctionnement du ventricule droit associé à une embolie pulmonaire. L’Impella est le seul dispositif thérapeutique cardiovasculaire ayant reçu l’autorisation d’utilisation d’urgence de la FDA pour traiter les patients atteints de la COVID-19.
________________
1 Lala et al. J Card Fail. 2018 ; 24 : 148-156
2 Mehta et al. J Am Coll Cardiol. 2001 ; 37 : 37-43
À PROPOS DES POMPES CARDIAQUES IMPELLA
Les dispositifs Impella 2.5® et Impella CP® bénéficient d’une approbation de pré-commercialisation de la FDA pour le traitement de certains patients atteints d’insuffisance cardiaque avancée qui subissent des interventions coronaires percutanées (ICP) planifiées ou urgentes, telles l’angioplastie par pose de stent ou de ballonnet, visant à rouvrir des artères coronaires obstruées. Les dispositifs Impella 2.5, Impella CP, Impella CP avec SmartAssist®, Impella 5.0®, Impella LD® et Impella 5.5® avec SmartAssist® sont des pompes cardiaques approuvées par la FDA utilisées pour traiter les patients subissant des crises cardiaques ou atteints de cardiomyopathie en état de choc cardiogénique. Elles possèdent la capacité unique de permettre la récupération de la fonction cardiaque native, permettant ainsi aux patients de rentrer chez eux sans être greffés. Le dispositif Impella RP® est approuvé par la FDA pour traiter l’insuffisance ventriculaire droite ou une décompensation suite à l’implantation d’un dispositif d’assistance ventriculaire gauche, un infarctus du myocarde, une transplantation cardiaque ou une opération à cœur ouvert. L’utilisation en urgence de la pompe Impella RP par du personnel soignant en milieu hospitalier est également permise afin de fournir une assistance ventriculaire droite temporaire pour une période allant jusqu’à 14 jours chez des patients en soins intensifs ayant une surface corporelle ≥ 1,5 m2, pour le traitement de l’insuffisance cardiaque droite aiguë ou de la décompensation causée par des complications liées à la Covid-19, y compris l’embolie pulmonaire. La pompe Impella RP n’a été ni autorisée ni approuvée pour le traitement de l’insuffisance cardiaque droite aiguë ou de la décompensation causée par des complications liées à la Covid-19. Les systèmes d’assistance ventriculaire gauche (VG) Impella sont également autorisés pour une utilisation d’urgence par le personnel soignant en milieu hospitalier pour fournir une décharge et une assistance VG (≤ 4 jours pour Impella 2.5, Impella CP, et Impella CP avec SmartAssist; contre ≤ 14 jours pour Impella 5.0 et Impella 5.5 avec SmartAssist) dans le traitement de patients en soins intensifs atteints d’une infection à la COVID-19 confirmée suivant un traitement ECMO et ayant développé un œdème pulmonaire durant une assistance ECMO V-A ou une décompensation cardiaque tardive à la suite d’une myocardite durant une assistance ECMO V-V. Les systèmes d’assistance VG Impella n’ont été ni autorisés ni approuvés pour l’indication d’utilisation autorisée. L’utilisation de la pompe Impella RP et des Systèmes d’assistance VG Impella a été autorisée par la FDA pour les cas d’urgence mentionnés ci-dessus, en vertu d’une autorisation d’utilisation d’urgence (EUA), et a été autorisée uniquement tant que la déclaration de l’existence de circonstances justifie l’autorisation d’utilisation d’urgence de dispositifs médicaux en vertu de la section 564(b)(1) de la loi, 21 U.S.C. § 360bbb-3(b)(1), à moins que l’autorisation ne soit résiliée ou révoquée plus tôt.
En Europe, les dispositifs Impella 2.5, Impella CP et Impella CP avec SmartAssist sont certifiés CE pour le traitement des patients ICP à risque élevé et des patients IAM en état de choc cardiogénique pendant une durée maximale de 5 jours. Les pompes Impella 5.0 et Impella LD sont certifiées CE pour le traitement des patients atteints d’un infarctus ou d’une cardiomyopathie en état de choc cardiogénique pendant un maximum de 10 jours. La pompe Impella 5.5 avec SmartAssist est certifiée CE pour le traitement des patients atteints d’un infarctus ou d’une cardiomyopathie en état de choc cardiogénique pendant une durée maximale de 30 jours. La pompe Impella RP est certifiée CE pour le traitement de l’insuffisance ventriculaire droite ou d’une décompensation suite à l’implantation d’un dispositif d’assistance ventriculaire gauche, un infarctus du myocarde, une transplantation cardiaque, une opération à cœur ouvert ou une arythmie ventriculaire réfractaire. Pour en savoir plus sur le portefeuille de pompes cardiaques Impella, y compris leurs indications approuvées et les informations importantes en matière de sécurité et de risque associées à l’utilisation de ces dispositifs, rendez-vous sur www.impella.com.
À PROPOS D’ABIOMED
Basé à Danvers, dans le Massachusetts, aux États-Unis, Abiomed, Inc. est un chef de file des dispositifs médicaux offrant une assistance circulatoire. Nos produits sont conçus pour permettre au cœur de se reposer en améliorant la circulation sanguine et/ou en assurant le pompage du cœur. Pour plus d’informations, veuillez consulter : www.abiomed.com. Abiomed, Impella, Impella 2.5, Impella 5.0, Impella 5.5, Impella LD, Impella CP, Impella RP, SmartAssist et Impella Connect sont des marques déposées d’Abiomed, Inc., et sont enregistrées aux États-Unis ainsi que dans certains autres pays. Impella BTR, Impella ECP, CVAD Study et STEMI DTU Study sont des marques commerciales en instance d’Abiomed, Inc.
DÉCLARATIONS PROSPECTIVES
Le présent communiqué contient des déclarations prospectives, notamment des déclarations concernant le développement de produits existants et de nouveaux produits d’Abiomed, l’évolution de l’entreprise en matière de croissance commerciale, les opportunités futures et les approbations réglementaires attendues. Les résultats réels de la société peuvent être sensiblement différents de ceux escomptés dans ces déclarations prospectives en raison d’un certain nombre de facteurs, notamment les incertitudes liées à la portée, l’ampleur et la durée de la pandémie de la COVID-19, au développement, aux tests et aux approbations réglementaires connexes, comme le potentiel de pertes futures, une fabrication complexe, des exigences de qualité élevées, la dépendance à l’égard de sources d’approvisionnement limitées, la concurrence, les changements technologiques, la réglementation gouvernementale, les litiges, les besoins en capitaux à l’avenir et l’incertitude quant à l’obtention de financements supplémentaires, ainsi que d’autres risques et défis détaillés dans les documents déposés par la société auprès de la Commission américaine des valeurs mobilières et boursières (SEC), notamment le dernier rapport annuel déposé sur formulaire 10-K ainsi que les documents déposés ou présentés par la suite auprès de la SEC. Le lecteur est prié de ne pas se fier indûment aux déclarations prospectives, qui sont valables uniquement à la date du présent communiqué. La société n’est aucunement tenue de publier les résultats d’une quelconque révision de ces déclarations prospectives qui pourraient être faites afin de refléter des événements ou des circonstances survenant après la date de publication de ce communiqué ou pour refléter la survenue d’événements imprévus.
Le texte du communiqué issu d’une traduction ne doit d’aucune manière être considéré comme officiel. La seule version du communiqué qui fasse foi est celle du communiqué dans sa langue d’origine. La traduction devra toujours être confrontée au texte source, qui fera jurisprudence.
Contacts
Tom Langford
Directeur de la communication
(978) 882-8408
TLangford@abiomed.com